FYB201
Ranibizumab-Biosimilar
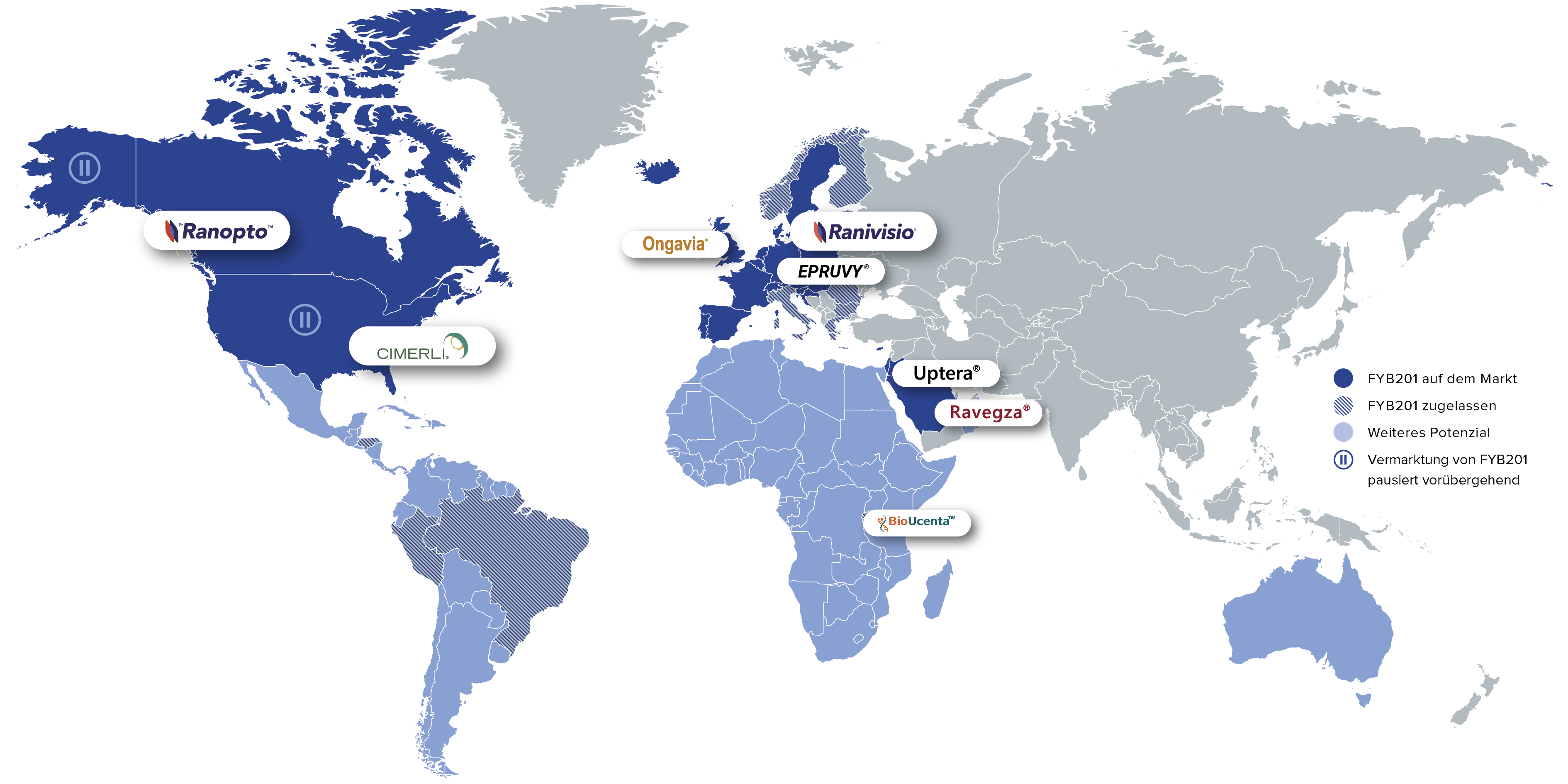
FYB201 ist als Behandlungsoption für Patientinnen und Patienten mit schweren Netzhauterkrankungen derzeit in 24 Ländern weltweit auf dem Markt. Weitere Produkteinführungen – unter anderem in Lateinamerika und auf dem afrikanischen Kontinent – sind geplant.
Indikationsgebiet
Ophthalmologie
Wirkstoffgruppe
VEGF-Inhibitoren
Indikationen des Referenzarzneimittels
Neovaskuläre (feuchte) altersbedingte Makuladegeneration, Diabetisches Makulaödem, Choroidalen Neovaskularisation, Proliferative diabetische Retinopathie, Makulaödem infolge eines Netzhautvenenverschlusses*
Markteintritt
2022
Business-Modell:
50 % Formycon-Projekt über die Beteiligung an der Bioeq AG, einem Joint Venture der Formycon AG und der Polpharma Biologics Group B.V.
Ranibizumab-Markt
Ranibizumab gehört zu den Standardtherapien im Anti-VEGF-Indikationsbereich. Im Jahr 2025 verzeichnete das Referenzarzneimittel Lucentis® einen weltweiten Umsatz von rund 600 Mio. US$.
Kommerzialisierungspartner:
![]()
Brands: Ranivisio®, Ongavia®, Ranopto®
Region: EU, Großbritannien, Kanada
![]()
Brands: Cimerli®, Epruvy®
Region: US, Deutschland
![]()
Brands: Nufymco®
Region: US
![]()
Brands: Ravegza®, Uptera®
Region: MENA
![]()
Brands: BioUcenta
Region: Sub-Sahara Afrika
![]()
Brands: Ranivisio®
Region: Brasilien
sind in der Produktinformation der EMA oder FDA zu entnehmen.
FYB201 Biosimilar-Entwicklung
FYB201 Biosimilar-Entwicklung
So wirkt FYB201 (Ranibizumab)
FYB201/Ranibizumab ist ein monoklonaler Antikörper, der zur Behandlung von Erwachsenen mit Sehbeeinträchigungen aufgrund einer Schädigung der Netzhaut – insbesondere der Makula – verabreicht wird. Die Makula sorgt für das Sehvermögen, das für alltägliche Tätigkeiten wie Autofahren, Lesen und Erkennen von Gesichtern erforderlich ist.
Der Antikörper bindet an den vaskulären endothelialen Wachstumsfaktor A (VEGF-A), der das Wachstum von Blutgefäßen fördert und zum Austreten von Flüssigkeit und Blut führt. Das ist die Ursache für die Schädigung der Makula. Durch die Blockierung von VEGF-A reduziert Ranibizumab das Wachstum der Blutgefäße, sowie das Austreten von Flüssigkeit und die Schwellung.
Klinische Daten aus der Phase-III-Wirksamkeitsstudie

Ranivisio®ist eine eingetragene Marke von Bioeq AG · Ranopto™ ist eine Marke von Teva Canada Limited
Ravegza® und Uptera® sind eingetragene Marken von MS Pharma