FYB201
Ranibizumab Biosimilar
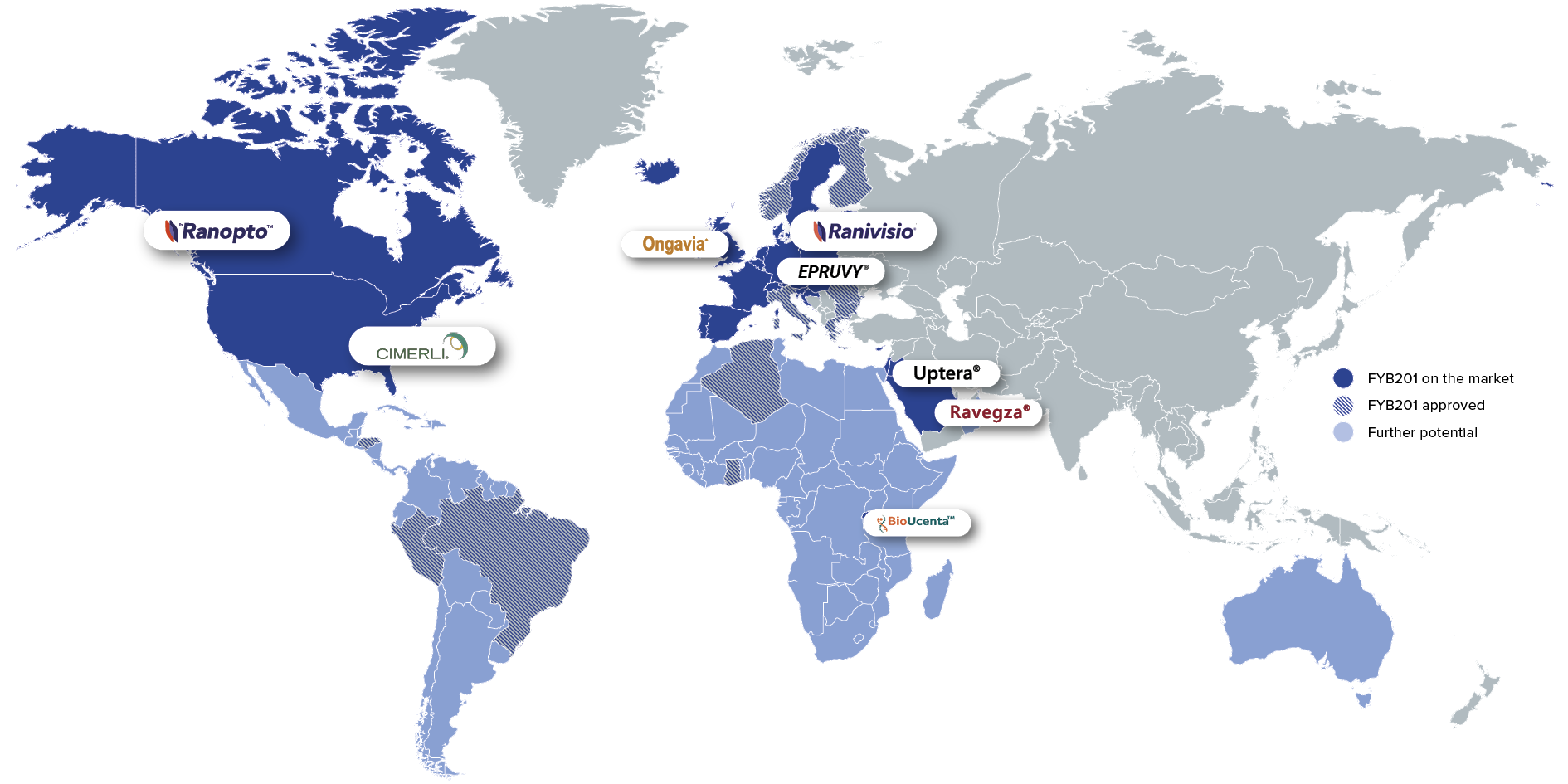
The Lucentis® Biosimilar FYB201 is currently available as a treatment option for patients with severe retinal diseases in 24 countries worldwide. Additional product launches – including in Latin America and on the African continent – are to follow.
Indication Area
Ophthalmology
Active Ingredient Group
VEGF inhibitors
Indications of the Reference Drug
Neovascular (“wet”) age-related macular degeneration (nAMD), Diabetic macular edema (DME), Choroidal neovascularization (CNV), Proliferative diabetic retinopathy (PDR), Macular edema following retinal vein occlusion (RVO)*
Market Launch
2022
Business Modell:
50% Formycon project via the stake in Bioeq AG, a joint venture of Formycon AG and Polpharma Biologics Group B.V.
Ranibizumab Market
Ranibizumab is one of the standard anti-VEGF therapies. In 2025, the reference drug Lucentis® generated global sales of approximately $600 million.
Commercialization partners:
![]()
Brands: Ranivisio®, Ongavia®, Ranopto®
Region: EU, UK, Canada
![]()
Brands: Cimerli®, Epruvy®
Region: US, Germany
![]()
Brands: Nufymco®
Region: US
![]()
Brands: Ravegza®, Uptera®
Region: MENA
![]()
Brands: BioUcenta
Region: Sub-saharan Afrika
![]()
Brands: Ranivisio®
Region: Brazil
provided by the EMA or FDA.
FYB201 Biosimilar Development
FYB201 Biosimilar Development
This is how FYB201 (ranibizumab) works
FYB201/Ranibizumab is a monoclonal antibody that is applied to treat adults with visual impairment due to damage to the retina, particularly the macula. The macula is important for the vision needed for everyday activities such as driving, reading and recognizing faces.
The antibody binds to vascular endothelial growth factor A (VEGF-A), which triggers the growth of blood vessels and leads to the leakage of fluid and blood. This is the cause of severe damage to the macula. By blocking VEGF-A, ranibizumab reduces the growth of blood vessels, fluid leakage and swelling.

Ranivisio® is a registered trademark of Bioeq AG · Ranopto™ is a trademark of Teva Canada Limited
Ravegza® and Uptera® are registered trademarks of MS Pharma